We are proud to present Aligned Elements V3.0, that includes:
Released Document View
Generate and Autopopulate Documents
An upgraded Review Experience
More than 250 new features and fixes
If you are interested in an in-depth demonstration of Aligned Elements, contact us on This email address is being protected from spambots. You need JavaScript enabled to view it..
The webinar can be viewed below:
Enjoy!
Written by Karl Larsson on . Posted in Product News.
By popular request, we're introducing a bundle of UI improvements to our Aligned Elements Users.
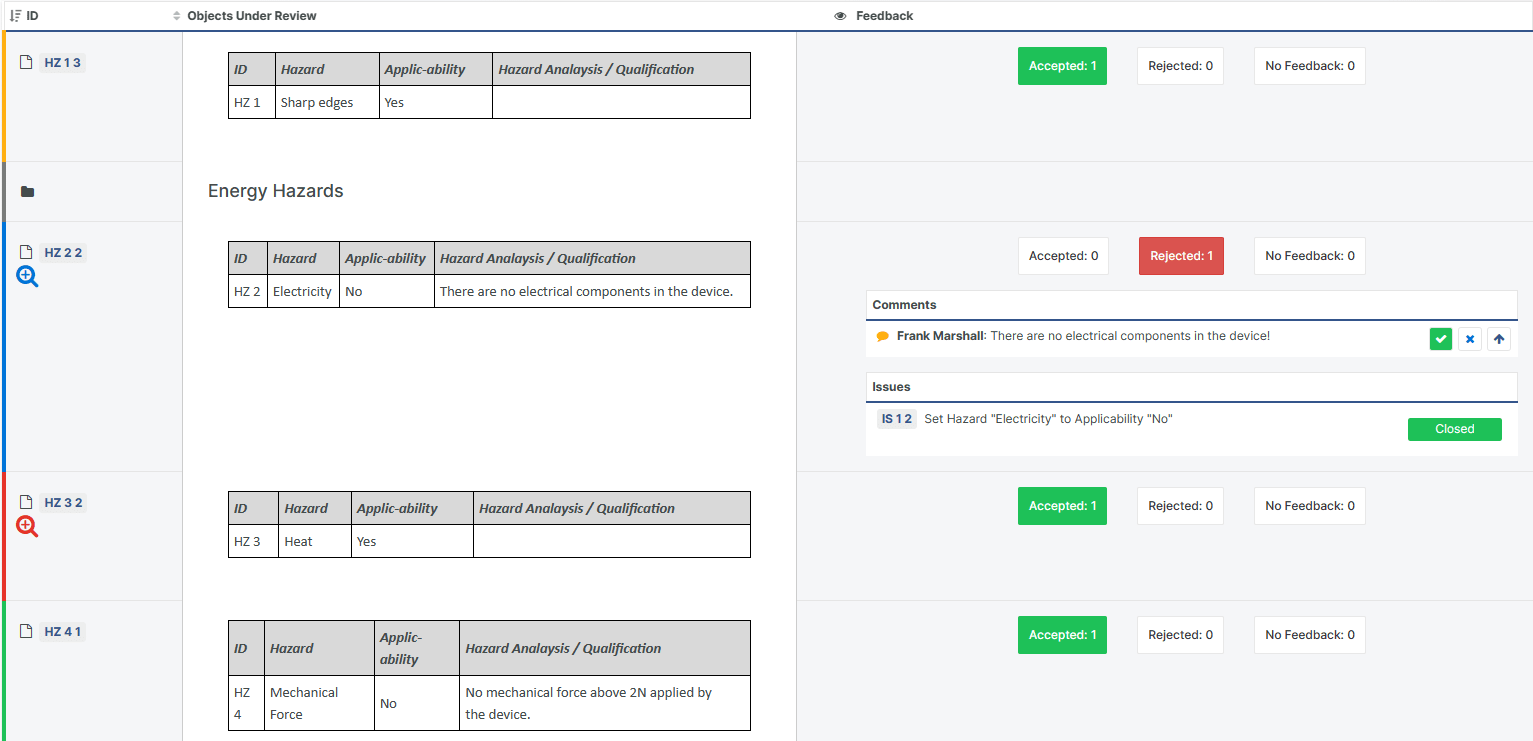
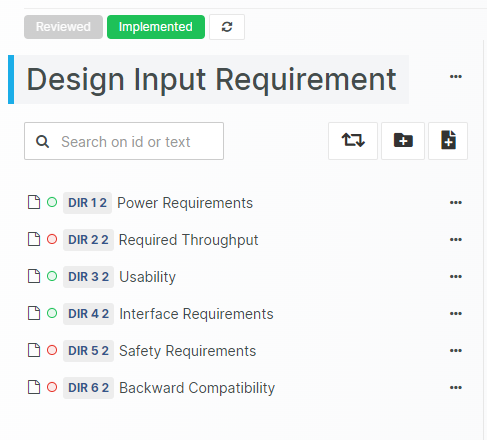
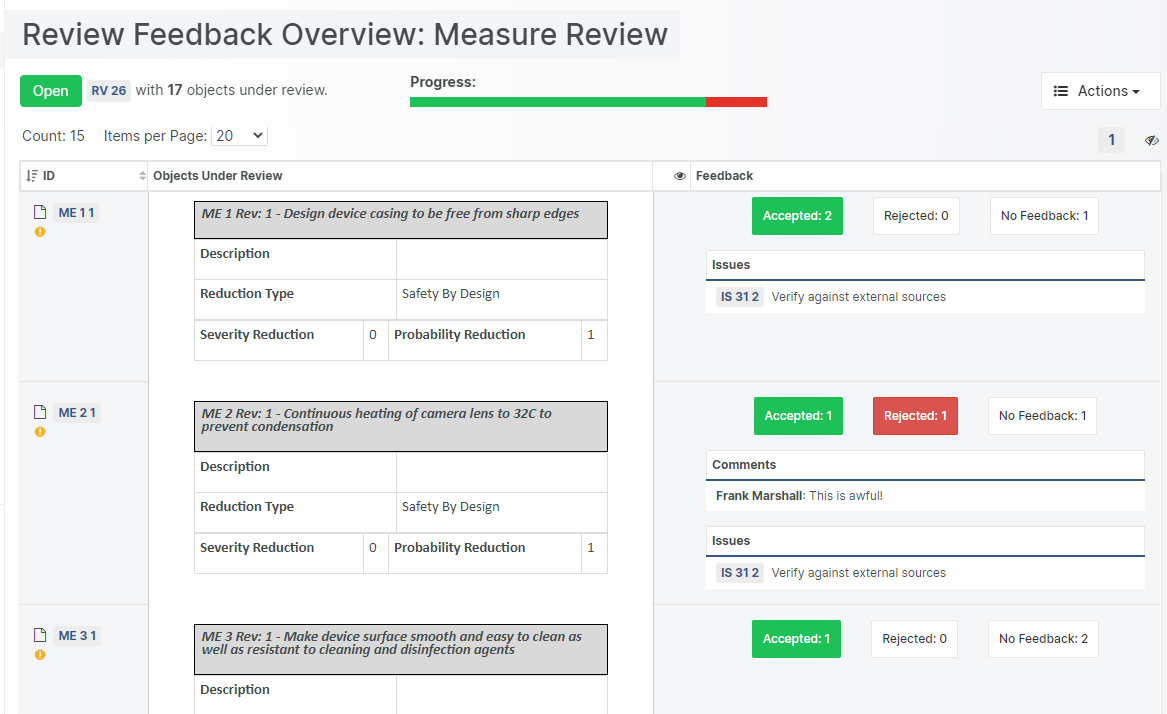
An Upgraded Review Experience
Our popular Design Review functionality permits online collaboration during the review process. Each user enters observations and feedback at their own pace, which later can be addressed in an organized fashion.
However, the Review process may differ from company to company and hence, the Aligned Elements community reached out to us with suggestions to strengthen the Design Review further.
In Aligned Elements V3.0, we have therefore added things, like:
Marking review Feedback Comments as "Resolved" or "Declined"
Alternative Review outcomes, such as "Rejected" and "Postponed"
Visually indicating whether an item has been changed during the Review
A detailed Word Gap Report showing all review changes for each item, with side-by-side colour-coded comparison
Displaying current Issue status in the Review Feedback and Overview Views
Auto-suggestion of Snapshot name for Review Snapshots
We are grateful for the input the Aligned Elements community has provided us with, that has given us an opportunity to provide a richer Review experience. Keep those suggestions coming!
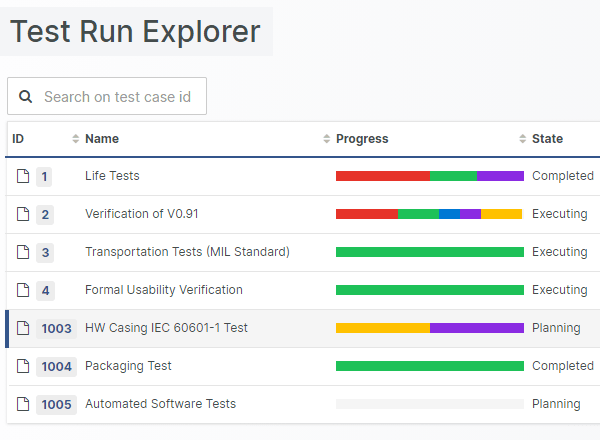
Test Run Management Performance enhancements
Testers tend to be the user group most intensely working with Aligned Elements. Therefore, giving them the best possible user experience is the least we can do to help. As a result, we have improved the Test Run and Test Run Explorer loading time, to speed up the switching between Test Runs, and Test Runs and Test artefacts.
We have also worked on speeding up the Trace setting mechanism, as well as cutting the execution times for queries and Word Report generation for artefacts with incoming traces.
New User Interface functions
We are excited to present a large number of new functions available in the Web Client User Interface. We hope that these will make the experience of Aligned Elements even more enjoyable.
Examples of these new features include:
Copy-paste of images from Word to Aligned Elements
Resize-ability of the Chapter View
Copy Chapter structures from one Design Control type to another
Edit File attributes in Display-As-List mode
Optionally refresh the Inconsistency Lists after resolving inconsistencies
Copying Design Control Items between Projects
Support for disabling Snapshots
Optionally rejecting in a Signature situation
These changes are available in the upcoming Aligned Elements v3.0 release.
Written by Karl Larsson on . Posted in Product News.
One of our best known features is Aligned Elements Word Integration.
This unrivalled feature lets you compose Design Control documents by filling your Word Templates with content from Aligned Elements.
You decide which data goes where and what the data should look like. The output is tailored to your company's process, product, templates and corporate design.
This is pure flexibility and adaptability, where you are in total control of the document creation.
When Control and Speed is all you need
However, there are situations where you don't want all that flexibility, cases when you need quite the opposite. Let's take the case of a Risk Assessment document. Your Risk Assessment word template has been meticulously crafted by your quality department. The template is set up to keep:
the Risk Identification list in section 3.2
the Risk Assessment itself in section 5
a Graphical Risk Summary in section 6 and
a list of residual risks in section 7.3.
All this data is available in Aligned Elements and your task is now to move it to the document itself.
The task is structured and clear. In this situation, flexibility and creativity is not necessarily what adds value.
Here our new feature come in to play.
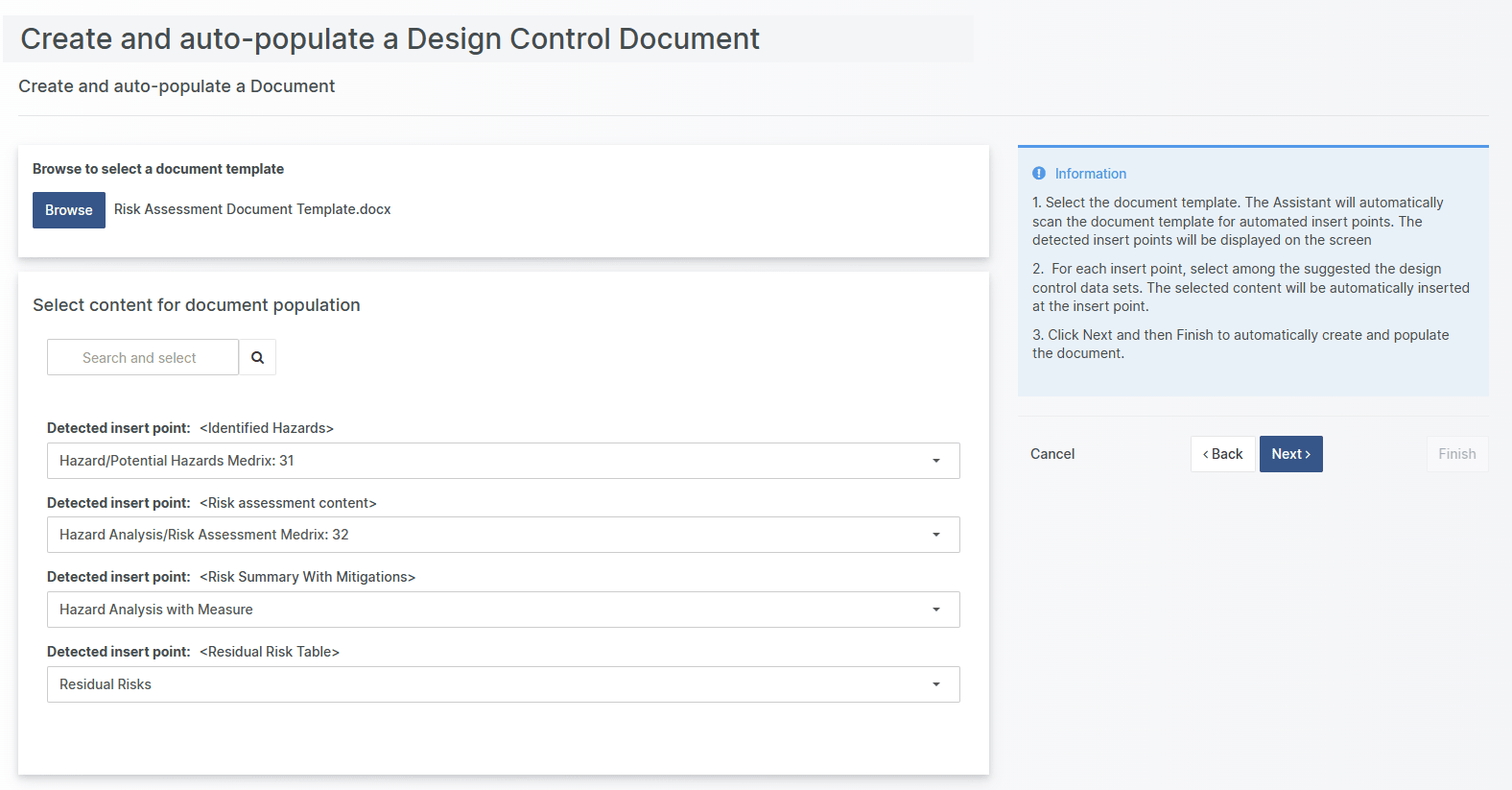
Introducing the Document Creation and Population Assistant
With Aligned Elements V3.0 introduction of the Document Creation and Population Assistant, you can now set up your Word Templates for automated creation and population.
In a few single clicks, the document is created and populated with Aligned Elements Design Control data without even having to open Word!
Select your word template in the Assistant and Aligned Elements will display the detected insert points. You can select among the data available in your project, or let Aligned Elements automatically suggest the data for your.
One click later, the document is created and populated automatically, becoming readily available in your Technical File.
The upsides for you are:
Less time spent on creating documents
Consistent Design Control placement in the documents
Less risk for human errors
Freeing up time for innovation
It has always been our goal to cut the time spent on medical device documentation. In our effort to free up even more time, we think we have come up with a helpful way to get to your documentation even faster.
This new feature is particularly interesting for medical device manufacturers that a rapidly expand their product portfolio or for projects with many re-occurring document types such as Test Case Reports, Review Protocols or Change Request documents. In other words, when many similar documents are frequently created, the Document Creation and Population Assistant is an excellent way to save time.
The Document Creation and Population Assistant is available in the Web Client of our upcoming Aligned Elements v3.0.
Written by Karl Larsson on . Posted in Product News.
The efficiency of your medical device company relies on your ability to manage and control the technical documentation.
These documents, being required by ISO 13485 and FDA QSR 820, must be correct, complete, and consistent.
To achieve this, solid document control is required.
However, systems and procedures that do not support efficient and streamlined document control can make the work overwhelming. When scrambling to get the Technical File ready for release, a stream of documentation activities occurs in parallel, with multiple updates and releases taking place frequently and simultaneously.
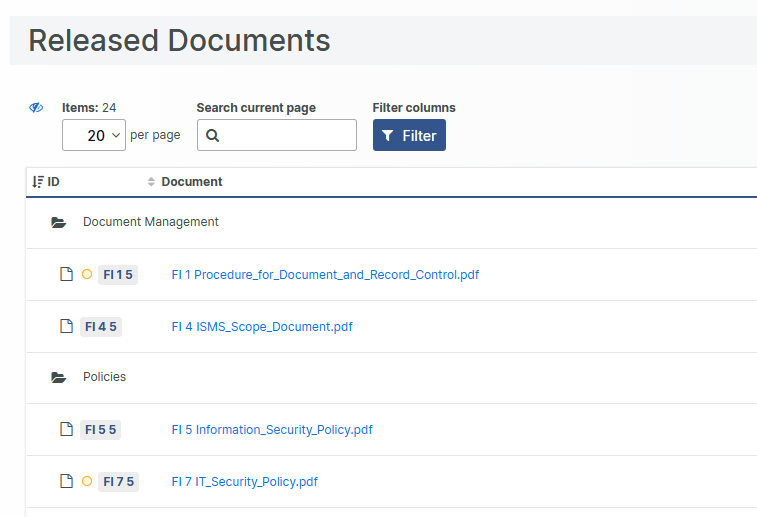
Released documents - up front
Attaining an overview of the currently approved, released state of all documents can be challenging and failing to do so, resulting in sending, or operating on, an outdated document version, can have significant effects.
For this reason, we announce the new "Released Documents" view, providing an unambiguous overview of the currently released documents in a single place. The documents displayed are your published, approved, and effective documents, in-short, your single source of (document-) truth.
Your team gets swift and exact access to the currently approved documents, appreciated during hectic audits and when scrambling to finalizing a submission. From now on, released documents are easily distinguishable from drafts, obsolete or superseded versions.
Powerful and flexible document management
The released state of your documents can be established with or without Aligned Elements built-in 21 CFR Part 11 compatible electronic signatures. Thus, you can decide yourself what makes a document being "released" in your Aligned Elements setup.
The released documents are organized and displayed in their corresponding file chapters for easier orientation. Additionally, visual indicators highlight documents where a new version is currently being drafted.
Filtering, searching, and sorting can be done using Aligned Elements standard mechanisms. In addition, Aligned Elements Tags can be applied to achieve quick filtering and grouping for selected purposes, such as all documents applicable to a standard or a geographical market.
Bulk operations such as download, analysis and report generation are available per default. Read-access to the "Released Documents" view is available to all users per default but can be displayed selectively by assigning the appropriate user permissions.
Edit Documents View - the broader and deeper access
The Released Documents view is the go-to place when looking for the currently approved and released version of a document.
However, for getting a broader and deeper view of the current work state, the "Edit Documents" view provides the tools you need. This view displays all documents in their live state, including unreleased documents in draft and ongoing updates of already released documents.
Conclusion
To provide a straight-forward and yet flexible Document overview that matches your organizations needs, this feature strengthens, streamline, and promote good document control practice in your Aligned Elements projects.
By integrating this new feature, you will achieve:
Direct and unambiguous entryway to released documents
Protection of documents from unauthorized access
Better and faster support for compliance activities
Improved document security
The Released Document view is available in the Web Client of our upcoming Aligned Elements v3.0.
Written by Karl Larsson on . Posted in Product News.
With the Hotfix Aligned Elements V2.6 Service Pack 2 (2.6.425/180.21104) we have fixed an important bug detected in V2.6 SP 1.
This bug fix concerns the trace direction of traces between Causes and Risk Analysis/Hazards Analysis objects in the Web Client, which were, at times, incorrectly set in V2.6 SP 1.
Aligned Elements V2.6 SP 1 (2.6.176/419.20790) is here and includes a number of bug fixes and a couple of new features.
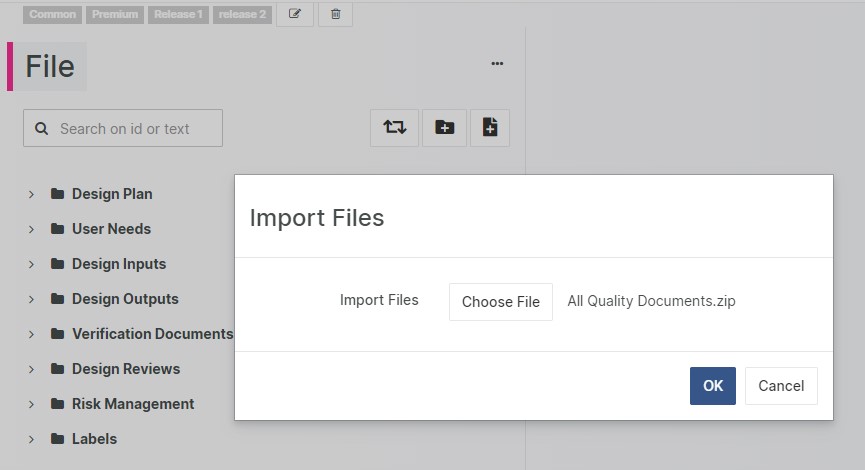
Upload Multiple Files
Perform bulk upload of files with a single click to Design Control types like Files and Attachments. Aligned Elements automatically creates the item, attaches the file, sets the title and recreates the chapter structure from the uploaded folder structure.
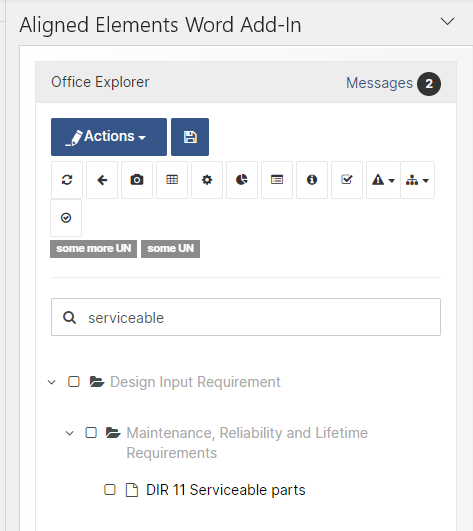
Search-as-you-type in Word Add-In
Locate your target Design Control Items quickly with search-as-you-type in the Aligned Elements Word Add-In.
Import Design Controls to selected Target Chapter
More control over your import actions are facilitated with the new option of importing Design Control Items into a selected chapter.
What's Changed
Aligned Elements Automator runs on both Linux and Windows
Customize wording for risk labels (Acceptable, ALARP, Inacceptable)
Improvements in Font and Formatting control
More control over Exported Excel Reports
Automatic email send out for signatures with enforced signing order
Prevent selected attributes to be copied at Generate
Display total number of Active Users
SQL Server 2022 compatibility
Upgrade now
With new features and important fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.6 SP 1 here.
Written by Karl Larsson on . Posted in Product News.
Aligned Elements V2.6 (2.6.157/401.20382) is here and includes new capabilities and numerous bug fixes.
Chapter Descriptions
Use Chapter Descriptions in Aligned Elements to add text displayed right below the Chapter when inserted in a Word Document.
Create rich Design Controls using Traced Object Attributes
The traced object attribute permits you to display data from traced objects in your Design Control. This can be singles Design Controls or lists of Design Controls, appear being embedded as attributes in your main Design Control.
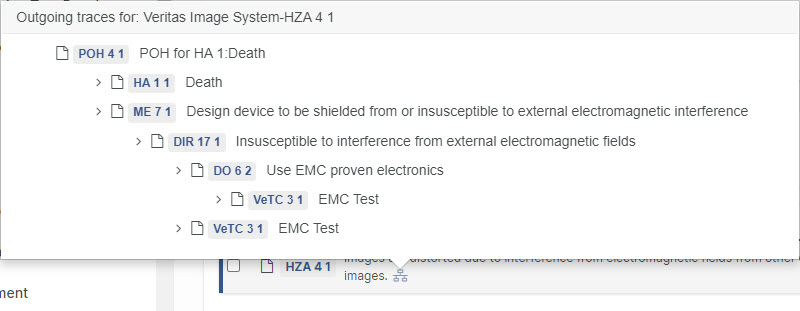
Inspect indirectly traced Design Controls
Hover with the mouse on trace inthe 'Trace To' and 'Trace From'-tabs to see indirect outgoing or incoming traces.
What's Changed
Trace Tables from Snapshot
Automatic Recovery of Unsaved Data
User Right for Test Run Management
Promote Comments to Issues in Review Feedback Overview
Trace Table allows "Upward" and "Advanced" at the same time
Export and import of Tags
File Object Revision History indicates Signed Documents
GemBoxPDF as new standard PDF Converter
Security Updates
Upgrade now
With new features and important fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.6 here.
Written by Karl Larsson on . Posted in Product News.
There are few standards that have higher influence on the daily workday than ISO 13485. Most processes used by a Medical Device manufacturer are mentioned and regulated by this ISO standard.
If you want to check how well your organisation fulfils the ISO 13485, you can use this new Aligned Elements Extension in your Aligned Elements instance.
This checklist extension covers key clauses in the sections:
With this checklist it is easier for you to assess what you have completed and what the remaining items are. Use the standard Aligned Elements functions to collaborate, analyse and report your results.
As a medical device manufacturer, you are probably well aware of the increasing focus on cyber security. When developing your device, you are responsible for ensuring that it is safe to use from a cyber security perspective. I think it is safe to say that the attention placed on cyber security aspects by autors and national authorities will only increase in the coming years.
FDA has issues a number of guidelines on how to implement and apply cybersecurity both on a process and product level. It is up to you as a medical device manufacturer to provide adequate cyber resilience in your products as well as the documentation required to prove it.
We are now happy to include the new OWASP Top 10 Checklist to our library of Cyber Security Extensions.
What is it?
OWASP stands for “Open Web Application Security Project”. It is a non-profit entity with international recognition and focuses on collaboration to strengthen software security around the globe.
OWASP publishes the OWASP Top 10 list, which provides rankings of, as well as a remediation guidance for, the top 10 most critical web application security risks.
It uses the extensive knowledge and experience of the OWASP’s open community contributors, and is based on a consensus among security experts from around the world. The OWASP Top 10 risks are ranked according to the frequency of discovered security defects, the severity of the uncovered vulnerabilities, and the magnitude of their potential impacts.
You can now easily assess and automatically document how well your medical device application stands up against the OWASP Top 10 by applying the Aligned Elements OWASP Top 10 Regulatory Assistant in your Aligned Elements projects.
How does it work?
This Assistant takes the shape of a Checklist where you assess your own medical device against the OWASP Top 10 security risks.
You start out by assessing whether the risk is applicable to your device at all, and if not, provide a qulified answer why this is not the case (the auditors rewards such qualifications).
If the risk is applicable, you are required to refer to the Aligned Elements Design Control Items that addresses the risk by selecting them in the UI. You can compare your risk reduction controls against known best-practice remidiations mentioned in the OWASP Top 10 list.
When the checklist is completed, a Regulatory Assistance item is generated, containing all steps and your provided answers. This information remains stored in Aligned Elements for compliance purposes.
The OWASP Top 10 Regulatory Assistant Checklist is free to all Aligned Elements customers and can be applied to any Aligned Elements Web Server installation.
Note! The OWASP Top 10 Regulatory Assistant Checklist only works in Aligned Elements.
Written by Karl Larsson on . Posted in Product News.
Aligned Elements V2.5 Service Pack 6 (2.5.144/373.19437) is here and includes new capabilities and numerous bug fixes
Drag and Drop Operations in Web Client
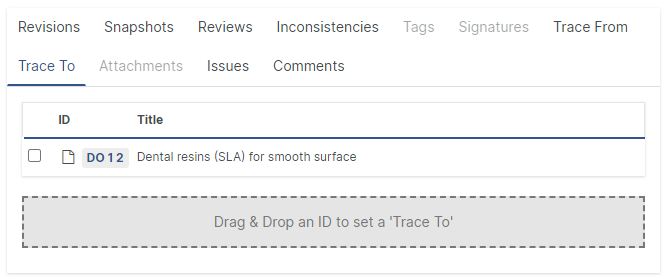
Setting incoming and outgoing traces and adding attachments is now possible using drag and drop, a faster and more intuitive way to work.
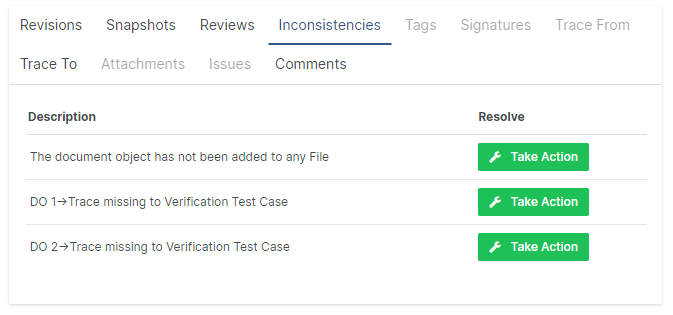
Fixing Inconsistencies on the spot
Unveiling inconsistencies and quality errors has always been one of the great advantages with Aligned Elements. Now, inconsistencies can be solved on the spot with just a few simple clicks. Aligned Elements offers a well curated selection of solutions to choose from, all adapted to your configuration, your access rights and the current project context.
Positive Cues - Indication of the Design Control's "health"
A Document Objects "health" is clearly displayed as a coloured dot in several list views. A Positive Cue is a collection of inconsistency rules that are evaluated together. The Positive Cue bar displays all Positive Cues available for the Design Control and also gives direct access to resolution options.
Regulatory Assistants
Run a Regulatory Assistant to quickly create or import Design Control items from predefined repositories and/or dynamically created items based on your user input.
What's Changed
Paste Images directly into Richtext and Table Attributes
Possibility to Map unknown import data to known project data
Assign User Groups as signees
Enforce order of Signing
User Management Enhancement
Allow adding new entries to Dynamic List Attributes on the fly
Two Factor Authentication in Web Client
Test Run: Batch Assign Users and Configurations to Test Cases
Upgrade now
With new features and important fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.5 SP 6 here.
Written by Karl Larsson on . Posted in Product News.
When you think about standards for medical device software, IEC 62304 is probably the first one that comes to mind. If you develop software for medical devices, this is definitely the standard you should look into.
Less known is IEC 62304's bigger brother, the IEC 82304 "Health software — Part 1: General requirements for product safety" which expands on the IEC 62304.
So what are the main differences? When shall I use which standard?
Differences between IEC 62304 and IEC 82304
Scope
IEC 82304 targets a larger range of device types than IEC 62304. IEC 82304-1 targets any kind of software, which directly or indirectly has an effect on health, known as "health software" in the standard. In contrast, IEC 62304 only targets software with medical intended use. Examples of device types that are in the scope IEC 82304 but not IEC 62304:
Radiology Information Systems
Prescription Management Systems
Laboratory Information Management Systems
Mobile Apps (not defined as FDA Mobile Medical Apps)
Standalone
IEC 82304 is intended for standalone software and not as IEC 62304, software embedded in medical devices or embedded in devices with specific hardware. Software running on PCs, Server or Mobile Devices on a general purpose Operating System are in the scope of IEC 82304.
Product Life Cycle Stages and Design Control Levels
As mentioned, IEC 82304 expands on IEC 62304 and addresses both additional Design Controls and requirements on expanded parts of the Product Life Cycle. It includes Product Use Requirements as well as Product Validation Plans and Reports. It specifies requirements on Product Identification and Instructions For Use as well as Post-market activities.
IEC 82304 references IEC 62304 for software development and maintenance, i.e. on a software level.
Aligned Elements IEC 82304 Configuration
The Aligned Elements IEC 82304 configuration is a superset of the IEC 62304 configuration. It expands the IEC 62304 configuration with Product Use Requirements and Product Validation Tests. It has been tuned to automatically take care of most of the involved quality checks, making sure that the required tasks and actions are sufficiently covered.
The Aligned Elements IEC 82304 configuration contains:
Pre-configured templates using IEC 82304 standard naming conventions
Software Safety Classification automatically based on risk analysis results
Numerous quality checks for consistency verification
Pre-configured Reviews and checkpoints according to IEC 82304 and IEC 62304 stipulations
Pre-configured Trace Tables based on the IEC 82304 and IEC 62304 requirements
A set of document templates being a great starting point for your documentation
48 importable Product Use Requirements for Accompanying Documentation from IEC 82304
The Aligned Elements IEC 82304 supports documentation management of:
Product Use Requirements
Product Validation Tests and Results
System and Software Requirements
Software Architecture building blocks(Software Items, Units, SOUPs, and segregations)
Risk Management using a Preliminary Hazard Analysis technique (listed in ISO 14971)
Software Verification (Unit, Integration and System testing)
Change and configuration management (Problem Reports and Change Management)
The Aligned Elements IEC 82304 Configuration can be downloaded here.
Written by Karl Larsson on . Posted in Product News.
ISO 14971 provides great assistance when identifying potential Hazards through its annexes, but it does lack a strong identification catalogue for Software Hazards.
Luckily, IEC TR 80002-1:2009 Annex B comes to rescue. This Technical Report, "Medical device software - Part 1 Guidance on the application of ISO 14971 to medical device software", takes a software angle on ISO 14971, just as the title suggests.
Annex B of the report elaborates on possible software hazards and factors to consider in order to properly assess whether and how they are applicable to a particular device.
To assist the risk identification of software hazards, we have built this extension, inspired by the Annex B of IEC/TR 80002-1.
It includes:
RVT file for a Software Hazard aspect and a corresponding DOCX Reporting style template
70+ importable potential Software Hazards to assess and integrate into your Risk Assessment
You may of course expand this hazard list with hazards that are particular to your device and the conditions under which it needs to operate.
This extension package can be combined with other risk identification packages from Aligned or in-house developed approaches by the manufacturer.
The "Software Hazard Identification based on IEC 80002-1 Annex B" extension is free to Aligned Elements users.
For more information on how to include the hazards in your risk assessment, contact the This email address is being protected from spambots. You need JavaScript enabled to view it. today.
Written by Karl Larsson on . Posted in Product News.
If you develop software for a medical device then you have hopefully aligned your processes with IEC 62304 - Medical Device Software.
In an auditing situation, you might be required to demonstrate how you comply to this standard.
We give you a fast and trackable way to do just that!
Our IEC 62304 Checklist is integrated in Aligned Elements and lets you perform the assessment inside AE, taking advantage of inconsistency rules, tracing to existing project documents as objective evidence and simple export to word once completed.
The checklist includes 93 prepared audit questions based on the requirements in IEC 62304.
This is a simple and efficient way to demonstrate your company and software's compliance with the standard.
In many products, software is an important component of medical devices. With increasing demands for greater functionality within medical devices, the complexity of medical device software development also increases. This, therefore, places increased demands for appropriate traceability.
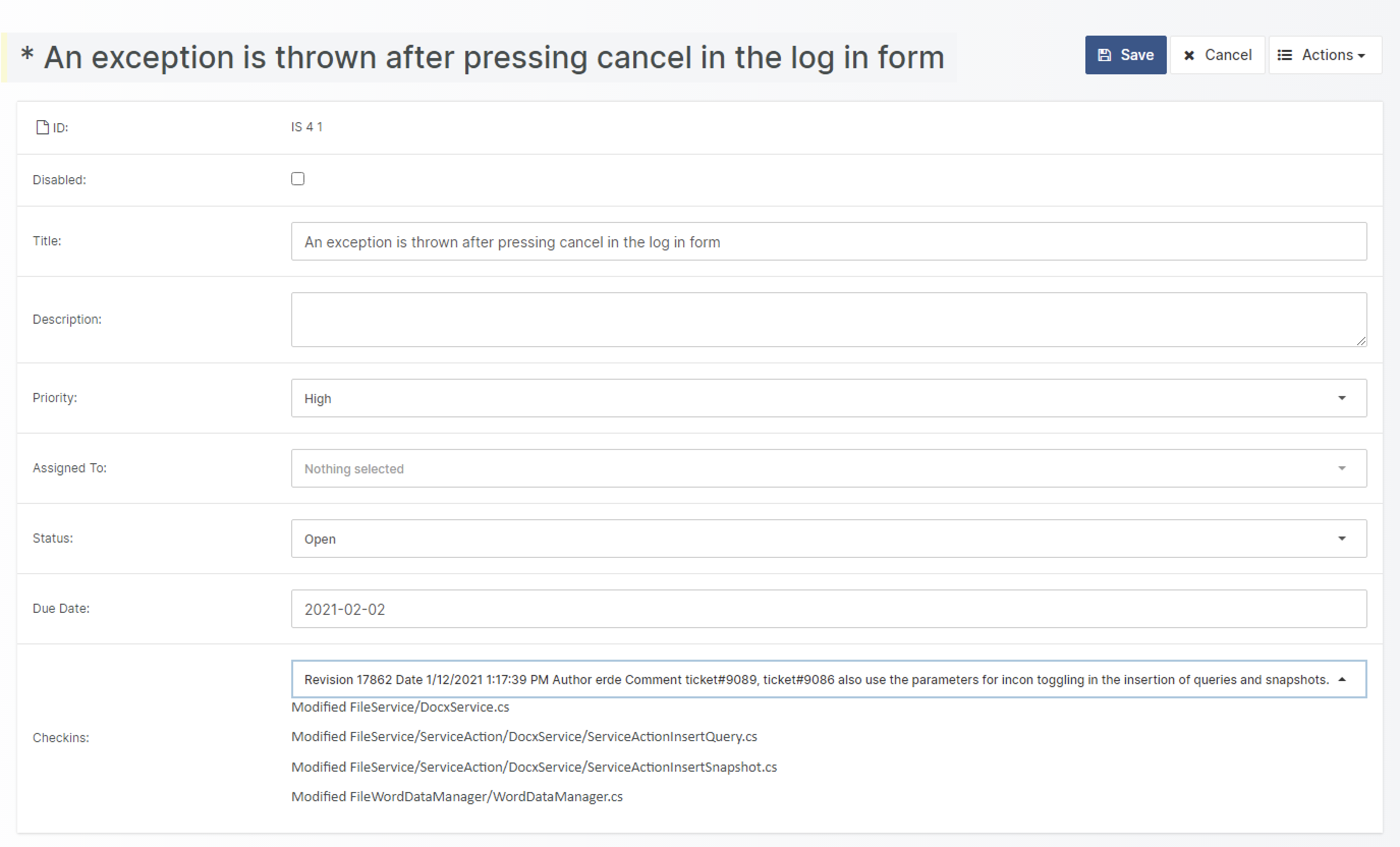
With Aligned Element V 2.5 SP 4, it is now possible to directly connect to your Subversion Repository.
This connection can be added as an additional field to any existing type.
A typical scenario for this would be to specify a specific check-in when closing an Issue. It is also possible to create a completely new type, for example, a “Source Code Item”. This type can then be included in the traceability landscape of your project.
Furthermore, the displayed source code files can be filtered by their file extension.
These features will allow you to:
Maintain traces between requirements, UML designs, test cases, and their associated source code.
Trace from Source Code to associated tests or reviews.
Through Inconsistency Rules, find out which UML designs or requirements have not been implemented yet.
When closing an issue, you can document in which SVN check-in the Issue has been addressed.
Easily create trace tables from any object to Source Code.
In reports, in addition to the name of the source code file, display the actual source code.
If you are interested in adding the Subversion Integration to your Aligned Elements project, please contact us at This email address is being protected from spambots. You need JavaScript enabled to view it.
Written by Karl Larsson on . Posted in Product News.
Aligned Elements V2.5 Service Pack 4 (2.5.344/120.17963) is here and includes some awesome new features and a host of bugfixes
Web Client UX Overhaul
A host of new UX features and an updated color scheme makes the use of Aligned Elements an even more efficient and pleasurable experience.
New Design Review Experience
Let each Review Team members inspect and provide feedback on Items under Review, aided by great analytics and feedback capabilities.
Highlight Inconsistent Items in Word
For easier inspection and faster reviews, automatically highlight inconsistent items in word documents to quickly visualize and address outstanding issues.
What's Changed
Integration with Subversion
Integration with GitLab
Configurable Email content
Update Fonts in items with Batch Change
Replace Words in items with Batch Change
Queries for External Issue items
Automatic Change Comments
New Inconsistency Rule: check for unreviewed items in Documents
New Inconsistency Rule: item must exist in signed Document
Warn when trying to edit item currently under Review
Group Attributes in Form
New Approval Attribute
Risk Summary Widget in Dashboard
Centralizing User Management in Web
Upgrade now
With exciting new features and important fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.5 SP 4 here.
Written by Karl Larsson on . Posted in Product News.
Have you ever struggled to describe Hazardous Situations so it was clear to all stakeholders what you intended to say?
Did you spend a lot of time to come up with a concisely written Sequence of Events and then the first person to review your document claims to not understand what you intended to convey?
When describing your harms, have you ever wished that someone had put together a list of all possible harms, so you could just pick the one, which is applicable for this particular situation?
And then after your product release, did a Risk occur that you did not foresee?
Common Terminology by curtesy of the IMDRF
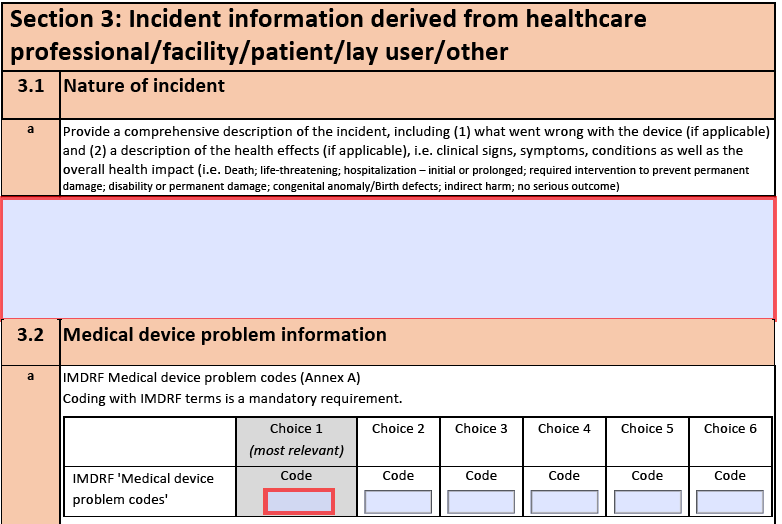
If you have ever experienced one or more of the above, there might be some help out there. The International Medical Device Regulators Forum (http://www.imdrf.org) has created a document called IMDRF terminologies for categorized Adverse Event Reporting (AER): terms, terminology structure, and codes.
Although that is quite a mouthful, this document can make your life a lot easier. It provides an extensive list of possible medical device problems, possible harms, and related causes. Each term is assigned a code, which has to be used when creating a Manufacturer Incident Report as required by the MDR (https://ec.europa.eu/docsroom/documents/41681).
Is the terminology only applicable for post-market events?
Although the terms compiled by the IMDRF have a strong focus on Post Market incidents, they are also useful in your pre-market design risk assessments. When performing your ISO 14971 compliant Risk Analysis during the development phase, a lot of time is (and should be) spent on the risk identification process to make sure all potential risks have been assessed and addressed.
In practice, this requires writing down and assessing the hazardous situations, what causes, and subsequent harms that could possibly arise by using your product. However, these are essentially the same as in a post-market scenario. Using the lists provided by IMDRF can speed up this process significantly.
So how does this make things easier for me?
The IMDRF lists act as an acceleration vehicle for your Risk Analysis. By using and analysing these established terms, you will save a significant amount of time when documenting all possible hazardous situations, causes, and harms. At the same time, the likelihood of overlooking a particularly hazardous situation, cause, or harm is greatly reduced.
Furthermore, ambiguities are reduced by using and referring to an established set of risk terminologies. Thus, you reduce the risk that other stakeholders, not just your colleagues, but also the auditors, will not misunderstand your carefully constructed Risk Analysis.
Using the IMDRF terminology in Aligned Elements
The lists are applicable to Aligned Elements projects using Risk Assessments using the Preliminary Hazard Analysis method.
It is possible to import the IMDRF items directly into Aligned Elements by using four import packages which you can download here.
The extension consists of lists containing a Design Control type called “IMDRF Item”, which have the attributes “Code” and “Definition”.
When importing them, you will need to map the types to types that exist in your configuration.
Note that the lists contain a large number of items that may not all be applicable to your particular device.
A pre-assessment step of the list content is therefore recommended before applying them to production projects.
The following mappings should be done.
“Annex A, Medical Device Problems” (469 items) should be mapped to a type that represents “Potential Hazards” in your configuration.
“Annex D, Investigation Conclusions” (35 items) should be mapped to a type that represents “Causes” in your configuration.
“Annex E, Health Effects - Clinical Signs and Symptoms or Conditions” (797 items) should be mapped to a type that represents “Harms” in your configuration.
“Annex F, Health Effects - Health Impacts” (64 items) should be mapped to a type that represents “Harms” in your configuration.
Please do not hesitate to ask for assistance at This email address is being protected from spambots. You need JavaScript enabled to view it..
Written by Karl Larsson on . Posted in Product News.
Risk Identification is an early and essential part of the risk management process and ISO 14971 requires us to make a complete risk assessment, to identify ALL hazards.
But, how do we know if all of the hazards have been identified? How can we prove this?
You could brainstorm or have a whiteboard session gathering ideas that pop up, but the only way to truly achieve confidence in your risk identification process is by using a structured approach.
There are several techniques available depending on the assessed source, including:
Assessing established potential hazards from internal records or published standards
Analysis of the manufacturer's experience with similar medical devices
Conducting a User Task Analysis on the user’s interaction with the device to uncover use errors
Assessing Field data and published incidents from similar devices in use
Assessing critical components for safe and effective use
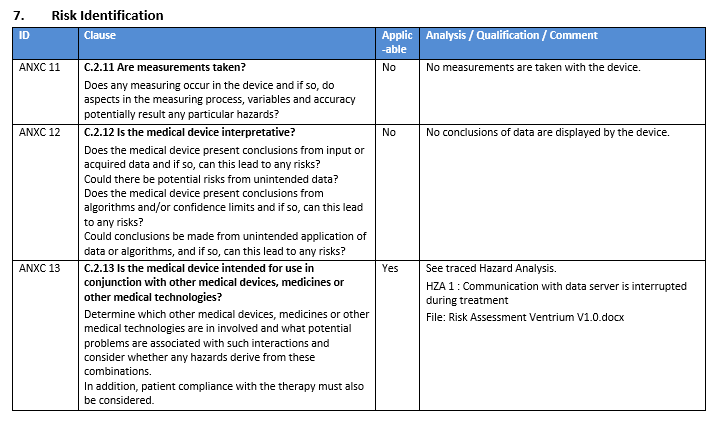
Because of the difficulty involved with thoroughly identifying all of the hazards, ISO 14971 provides a number of aides – such as Annex C (2012) (becoming the ISO 24791 Annex A in the 2019 edition) – which provide a list of questions to assist in establishing device characteristics that may impact safety. Although not exhaustive, these questions can serve as a starting point and become one of several potential approaches from which the complete risk identification can be assembled.
Aligned Elements users can kick start their risk identification process by downloading and importing our ISO 14971:2012 Annex C Extension, assessing them and start generating risks and mitigation.
The ISO 14971:2012 Annex C Extension contains:
RVT file for an ISO 14971 Annex C Question and a corresponding DOCX Reporting style template
37 importable questions built on Annex C in ISO 14971 to assess and integrate into your Risk Assessment
This Extension facilitates the assessment of the questions, the creation of both an automated assessment report of the Annex C questions as well as a starting point for generating new risks and mitigation.
It gives medical device manufacturers a predefined starting point when setting up their technical file with the intention of accelerating the documentation effort.
The user is of course welcome to expand this question list with questions that are particular for his/her device and the conditions under which it needs to operate.
The ISO 14971:2012 package can be combined with other risk identification packages from Aligned or in-house developed approaches by the manufacturer.
The ISO 14971:2012 Annex C package is free to Aligned Elements users.
For more information on how you can include our ISO 14971 questions in your risk assessment, contact the This email address is being protected from spambots. You need JavaScript enabled to view it. today.
Written by Karl Larsson on . Posted in Product News.
Aligned Elements V2.5 Service Pack 3 (2.5.313/114.16963) is here with a great improvement and some important fixes.
What's New
Collaborate using Comments
Use the Comments tab to add comments and send notifications to your team members. Comments can be used to discuss and clarify details or a particular design item during its life cycle. Emails notify comment authors about replies, upvotes, and mentions. Comments are only available in the Aligned Elements web client.
What's Changed
Important fixes regarding risk handling, Word, and merging
Upgrade now
With important improvements and a handful of fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.5 SP 3 here.
Written by Karl Larsson on . Posted in Product News.
Aligned Elements V2.5 Service Pack 2 (2.5.307/109.16875) is here and includes several integration improvements and a number of fixes.
Enterprise Architect Integration
Leverage the powerful System Integration models of Enterprise Architect in your Aligned Elements traceability! Enterprise Architect Diagrams can now be part of Aligned Elements item through a live connection with the Enterprise Architect repository enabling both traceability between Design Controls in Aligned Elements and models in Enterprise Architect diagrams as well as the incorporation of EA diagrams in Aligned Elements bi-directional Word reports.
Integration with Redmine
Use Aligned Elements integration with the popular Redmine ticket system to integrate Redmine tickets into your Design Control traceability. Create Redmine tickets from Aligned Elements on the fly when bugs are found during test execution, let Aligned Elements inconsistency checks validate the state of your Redmine items, and use Aligned Elements bi-directional Word Integration to incorporate Redmine ticket into your DHF documents.
Web Client Improvements
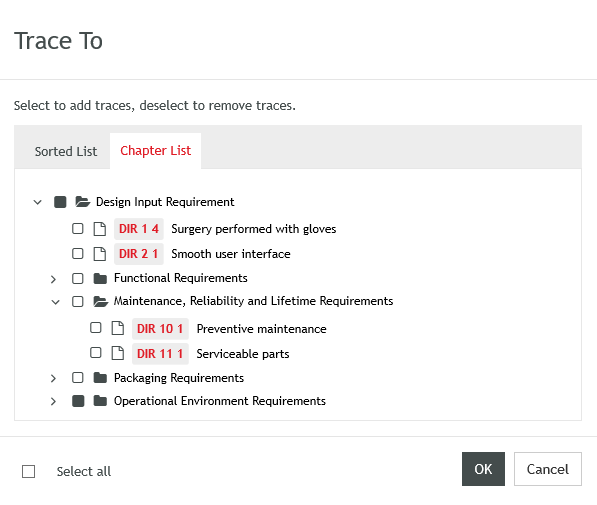
We have made a number of usability improvements available in the Aligned Elements Web Client to make your work easier and faster. These include displaying chapter structure in Trace Dialogs, one-click 'Add to Review' action from several views, single-click 'Save document as PDF', 'Find In...' functions to locate Design Control Items in different contexts, Excel exports from tables, and much more.
What's Changed
Integration with Perforce Jobs
Use Test Runs without Configurations
Single-click to find items in Trace / Project Explorer, Files and Test Runs
Compare Word documents using Word's Document Comparison feature
Save Files in their folder structure to disk
Convert files to PDF at save to disk
Do risk reduction in Hazard Analysis and not in Mitigations
Make Unlock Objects a separate user right
Save tables to Excel (Web client)
Revert to Revision (Web client)
Save Trace Table as word template (Web client)
Add to Review from several views (Web client)
Display items in chapter structure in Trace dialog (Web client)
Create Harms and Mitigations on the fly (Web client)
Optional mail invitations sent from Review and Signature (Web client)
Support for table operations in Table attribute (Web Client)
Upgrade now
With important improvements and a handful of fixes, this release is a recommended upgrade. Find the installer to Aligned Elements V2.5 SP 2 here.
Written by Karl Larsson on . Posted in Product News.
Sometimes I get the question "So how much time and money will I save by using Aligned Elements?"
A fair question and a question I can answer with a clean conscience: on average, you will save 30% or more of the time you currently spend on design documentation.
However, some medical device manufacturers can save (and have saved) a lot more than that!
If a modular documentation approach is applied, where reuse of already existing Design Control data is leveraged, there are examples of savings of up to 80%.
In this video below, you can find out more about the possibilities, prerequisites, and benefits received from Modular Design Documentation with Aligned Elements.
Happy Holidays!
Written by Karl Larsson on . Posted in Product News.