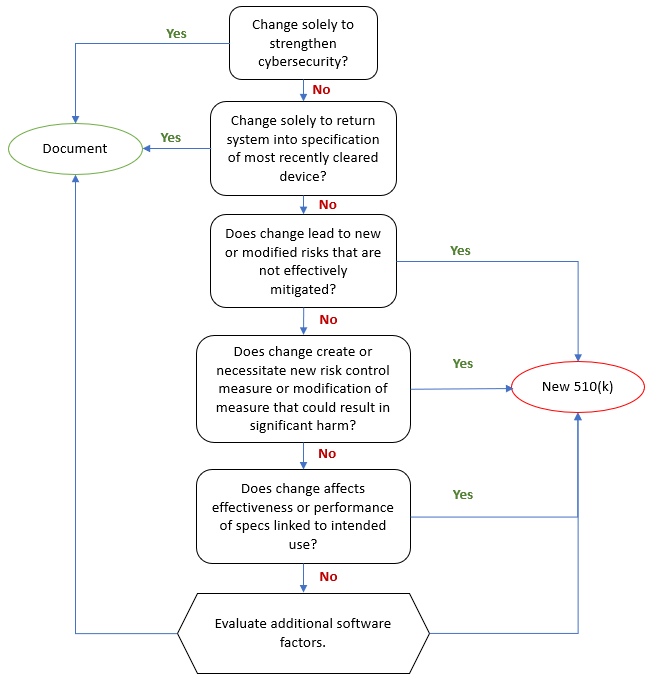
The medical device as a stand-alone product is a waning concept.
The desire to make use of the information collected in a medical device in other health systems, coupled with recent advancements in networks and interconnectivity has resulted in more devices being connected to the Internet. As a consequence, they have become more vulnerable to hackers.
A 2015 KPMG survey found that 81 percent of health care organizations had their data compromised within the previous two years.
FBI reports (FBI Cyber Division PIN (Private Industry Notification) #140408-010) that due to the transition of paper to electronic health records, lax cybersecurity standards and higher financial payout for medical records in the black market, the "cyber actors will likely increase cyber intrusions against health care systems".
During 2016, several ransom-attacks were launched against health institutions in the United States with wide-ranging consequences.
FDA has shown heightened interest in cybersecurity issues and released three guidelines during the last two years. Medical Device manufacturers are likely to focus more on cybersecurity risk management activities in the near future and assign additional resources accordingly.
One integral part of the cybersecurity risk management process constitutes the risk assessment.
Performing risk assessments is a core activity in the medical device industry and many of the available techniques are well-known and well-used by industry professionals. Having long and thorough experience of risk management might lead the same professionals to believe that cybersecurity issues can be harnessed by the tools already at hand.
There are, however, key aspects where cybersecurity risks differ from traditional medical device risks.
Risk identification and the building blocks of a cybersecurity risk
The fundamental challenge during risk identification is to ensure that all relevant risks have been identified.
Verifying that this criterion has been fulfilled is often difficult and therefore structural help and practical advice is very welcome.
Due to the wide variety of possible medical device types, ISO 14971, the standard for medical device risk management, understandably has a hard time defining concrete identification techniques that are relevant enough to provide value for every kind of medical device.
IT risk management, operating in a narrower technical scope, provides a host of techniques, tailored to this domain. Many of these techniques are based around the asset-vulnerability-threat model.
These components can be described as followed.
- Assets are the entities a cyber-intruder attempts to access and control. An asset has a value to the patient and therefore also to an intruder. In a safety-related context, we can exemplify assets as device configurations, health data, medical device functions, or battery power.
- Vulnerabilities represent weaknesses in the medical device that, when exploited, can give an intruder access to an asset. Software bugs, application design flaws, and insufficient input validation are examples of software vulnerabilities. But vulnerabilities can also be found in hardware, business processes, organizational structures, and interpersonal communication.
- A threat is defined as an event with the potential to an adverse impact on an asset. The threat is executed by a threat agent (i.e. an intruder) exploiting a vulnerability in order to access an asset.
A combination of these building blocks describes a cybersecurity risk.
As a starting point, the manufacturer should be able to enumerate assets for the given device. By analyzing how these assets can be targeted, e.g. by performing a “Threat modeling analysis”, using a data flow analysis of the application, identifying "data-at-rest" (data storage) and "data-in-motion" (data transfer) will further help the manufacturer to identify vulnerabilities and threats, particular to the medical device.
Bottom-line: Identifying assets, threats, and vulnerabilities will support the manufacturer when enumerating potential cybersecurity risks. The identified items should be listed in the risk management documentation.
What is the probability of a cyber attack?
The Medical Device industry takes its risk assessment cues from ISO 14971, which defines risk as the combination of the probability of the occurrence of harm and the severity of that harm.
The computer security industry, on the other hand, has used several kinds of assessment methods to estimate the "riskiness" of cybersecurity risks. None of them rely solely on the probability and severity of an event. Instead, these techniques estimate and/or quantify aspects of the assets, threats, and vulnerabilities that in combination say something about the computer security risk.
So how can the medical device risk assessment methods concerned with the safety of patients be connected with techniques whose primary concern is information security?
In the AAMI paper "TIR 57 - Principles for medical device security - Risk Management", the authors address this discrepancy and attempt to transpose the cybersecurity line of thinking into the domain of ISO 14971.
It is easy to see how the "potential adverse effect" can be regarded as analogue to the “Harm” in ISO 14971 and be quantified with a corresponding severity.
The probability factor in ISO 14971 does not have a direct equivalent in the cybersecurity risk domain. A composite factor called "exploitability" combining characteristics of the vulnerability, the threat agent, and the medical device itself is mentioned in the FDA guidelines. This factor intends to indicate the amount of work involved in order to invoke a successful attack.
The AAMI TIR57 group suggests a two-pronged approach to establish something similar, combining two likelihood factors.
The first factor, the “Threat Likelihood”, defines the likelihood of the threat agent having the motivation, skills, and resources to exploit a given vulnerability.
The second factor estimates the likelihood of harm being the effect of an exploited vulnerability. These two factors in combination make out the probability of the cybersecurity risk.
Note that this approach is similar to the P1 (probability of the hazardous situation occurring) / P2 (probability of the hazardous situation causing harm) frequently used in the medical device community.
Information security theory recognizes that aspects and characteristics of the threat agent, the vulnerability, and the device itself drive these factors.
Bottom-line: caution shall be taken when estimating probability/likelihood/exploitability for cybersecurity risks. The manufacturer's Risk Management SOP shall address this concern accordingly and be adapted if necessary.
The drivers of cybersecurity risks
During classic medical device risk assessments, the identified causes leading to hazardous situations and potentially to harms are in most cases accidental. Although malicious use should be considered, this area often receives considerably less attention than harms caused by accidental events.
For cybersecurity risks, the opposite is true. Here, a significant factor in the causal chain constitutes the intentional behavior of a threat agent causing harm by exploiting a vulnerability.
Whereas the software flaw might have been created accidentally, exploiting it is an intentional act.
A malicious agent may come in many shapes and forms, such as a criminal organization, a competitor, or disgruntled employees. Each of these has its own motivation, skill set, and reach when it comes to detecting and exploiting vulnerabilities, which affects the likelihood of a vulnerability being exploited as we have seen above.
Therefore, not only needs the focus to be shifted from accidentally caused risks to intentionally caused risks. The manufacturer benefits from analyzing the characteristics of the malicious intruder in order to do a proper risk assessment.
Bottom line: Manufacturers will need to shift perspective from accidentally caused risks to explicit maliciously caused risks during the identification and classification of the risk assessment.
The poisoned SOUP
Just like in the rest of the software industry, medical device companies use third-party libraries to increase productivity when developing medical devices.
These third-party libraries (or frameworks or applications), sometimes referred to as "SOUP" components (Software of Unknown Provenance) are of course also subject to cybersecurity scrutiny.
It is debatable if third-party components, and in particular open source components, are inherently more unsafe than proprietary produced applications (there are several arguments against such claims).
Regardless of this claim, it can be safely assumed that many of these libraries were not developed with a medical device cybersecurity context in mind. The security firm “Contrast Security” reports that 26% of downloaded SOUP:s contained known vulnerabilities and were still applied.
It shall be noted that SOUP:s themselves often rely on and integrate third-party software, which increases the scope of the problem accordingly.
The medical device manufacturer is responsible for any vulnerabilities caused by SOUP:s in his device and must therefore analyses his SOUP:s accordingly.
This shall include a systematic inventory of SOUP:s, actively collecting information on current vulnerabilities in SOUP:s as well as applying timely updates of the SOUP:s when available.
Bottom-line: The manufacturer must have a clear plan for how to handle potential cybersecurity risks in SOUP:s.
Skills required to assess cybersecurity risks
Cybersecurity vulnerabilities are largely made up of software flaws. Understanding the technical details of how such vulnerabilities come into being, the technical influence they have, and how they can be mitigated requires precise software knowledge. Including software developers in the assessment group is, therefore, a highly recommended measure.
It is a misconception that all software professionals are software security professionals. The majority of today’s information security problems can be traced to flaws in code. Many software developers lack basic training and understanding of cybersecurity.
As already mentioned, the security scope for an interconnected medical device is much larger than the device itself. The network infrastructure in which connected medical devices operate is often outside the control of the medical device manufacturer but still has a great impact on the overall risk exposure and needs to be included in the risk assessment.
It is likewise a misconception that cybersecurity is a strictly technical field. Cybersecurity vulnerabilities are not exclusively found in hardware and software but also in business processes, organizational structures, human behavior, and the environment in which the device operates. A thorough understanding of where, how, and by whom the medical device will be operated is therefore important input for the risk assessment.
Last but not least, the clinical experts are required for estimating the potential harm of compromised availability, confidentiality, and integrity of the associated data.
All these competencies are required in order to perform a comprehensive cybersecurity risk assessment and it is clear that it stretches beyond being an internal R&D activity.
The risk assessment benefits from involving stakeholders beyond the immediate software development team. If specific cybersecurity expertise is lacking in the organization, the manufacturer should consider employing or train their own experts or outsource these functions to a competent partner.
Bottom-line: the knowledge required to perform a medical device cybersecurity risk assessment is both broader and deeper than what is often immediately available in many medical device companies.
Conclusion
The medical device industry has extensive experience with risk management and risk assessment techniques.
The cybersecurity dimension of medical device development is an additional aspect where risk assessments can enhance safety.
However, traditional medical device risk assessment techniques need adaptation in order to successfully be applied to cybersecurity risks.
Cybersecurity risks consist of other aspects and other drivers than "classic" medical device risks and are best mitigated by recognizing these differences.
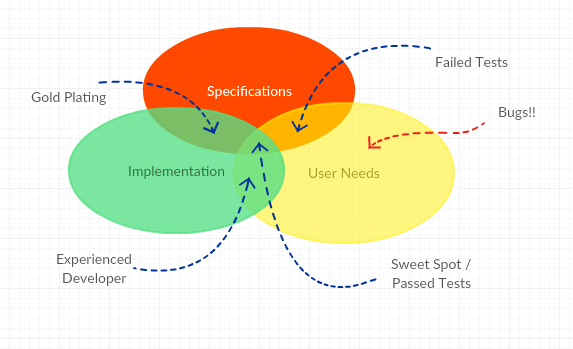
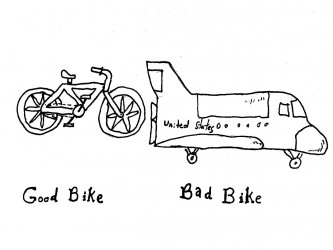 It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.
It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.