Software Change? Yes! - New 510(k)? Eh...
It is a well-known tendency that changes made to software (new features, bug fixes, etc.) often lead to new software errors. In a medical device context, this can have detrimental consequences.
The FDA requires that medical device manufacturers submit a new 510(k) when a marketed device has changes, including changes to software, that could significantly affect the safety or effectiveness of the device or when there are major changes in the intended use of the device.
Of course, the word "significantly" is the moving target here. To facilitate the decision whether a new 510(k) is required, the FDA issued the guidance document "Deciding When to Submit a 510(k) for a Software Change to an Existing Device" in October 2017.
FDA Commissioner Scott Gottlieb said a statement that the guidance ..." enhances predictability and consistency for innovators deciding when to submit new 510(k)s by better describing the regulatory framework, policies, and practices underlying such a decision.”
Aligned has published an Aligned Elements Regulatory Wizard called When to Submit a 510(k) for a Software Change to an Existing Device that codifies this guidance document and makes it easy to assess and document whether the software changes (captured and managed in Design Control Items in Aligned Elements) will result in a new 510(k) or if documentation therefore suffices.
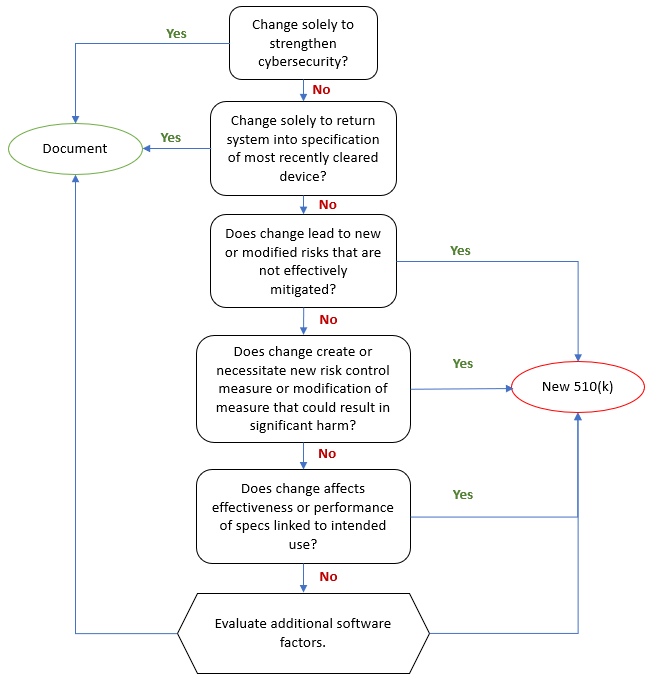
The wizard When to Submit a 510(k) for a Software Change to an Existing Device is freely available for download from the Extension section of this website and serves as yet another example of how Aligned Elements can reduce the development documentation effort of your device.
 ";
";