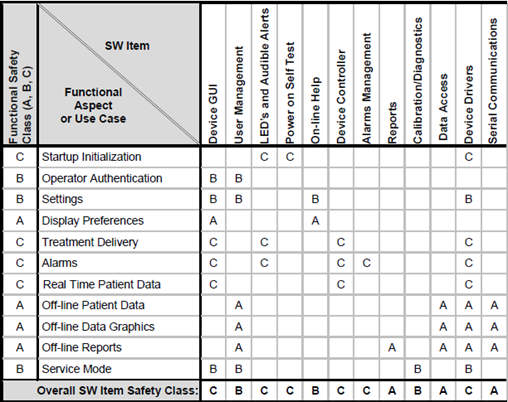
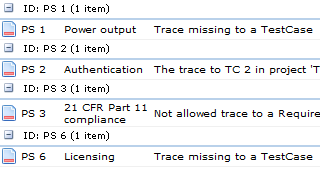
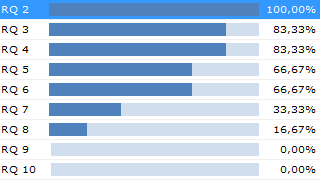
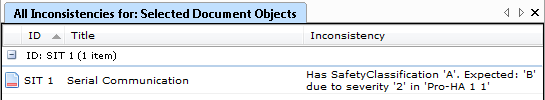
Innerhalb der Medizintechnik wird die Risikoanalyse, sei es in Form einer FMEA oder als vorläufige Gefahrenanalyse (PHA), zu einem immer wichtigeren Teil der Dokumentation. Sie spielt eine Rolle bei der Umsetzung von EN ISO 13485, IEC 62304, Richtlinie 93/42 EWG über Medizinprodukte und Richtlinie 98/97 EG über In-vitro-Diagnostika, sowie auch der Umsetzung anderer Normen und Direktiven.
Um schneller und effizienter entwickeln zu können, setzen immer mehr Hersteller auf eine modulbasierte Entwicklung. Um beiden Zielen gerecht zu werden, bietet es sich an, auch das gesamte Risikomanagement modular zu gestalten.
Das Zusammenspiel der Themen:
- Effizienz durch Wiederverwendung
- Konsistenz durch Vermeidung von Redundanz
- Organisatorische Herausforderungen
ist aber durchaus komplex.

Die Herausforderungen und ein möglicher Lösungsansatz werden in einem Gratis Paper hier skizziert.
{fastsocialshare}