If you're a medical device manufacturer preparing for MDR compliance or FDA approval, chances are you've already wrestled with the beast known as the Technical File. Whether you're aiming for a CE Mark, submitting for FDA Approval, or maintaining your Design History File, the stakes are high, and the pitfalls are real.
This article breaks down the five most common problems manufacturers face when compiling their Technical Documentation, and offers practical, actionable advice to help you avoid them. Based on insights from top regulatory consultants and notified bodies, this guide is designed to be more helpful than the usual checklist, style posts.
Let’s dive in.
1. Inadequate or disorganized Documentation
One of the biggest traps manufacturers fall into is assuming that “having the documents” is the same as being compliant. In reality, regulators don’t just care that you have the information, they care that it is complete, consistent, and logically structured. You might have all the right content, but if it’s buried in a maze of folders or inconsistently formatted, it becomes a problem.
Imagine an FDA reviewer trying to confirm your device’s intended use only to find three slightly different versions across three separate documents, or a notified body assessor giving up after scrolling through 1400 pages of an unindexed PDF.
Notified bodies and FDA reviewers expect clear, organized, and searchable documentation. For MDR, Annex II and III explicitly require this, and reviewers won’t dig through your files to find what they need.
Common symptoms:
- Missing or inconsistent device descriptions
- Poor document version control
- Scattered or duplicated information / documents
Fix it:
- Use a structured STED, check with your Notified Body if they recommend a particular one
- Implement document control systems with traceability
- Use a Design Control tool to manage reoccurring texts, such as Intended use and other parts of the Device Description
Remember: Your Technical Documentation isn’t just a formality, it’s your regulatory proof of safety and effectiveness. If reviewers can’t follow the story, they won’t sign off on the ending.
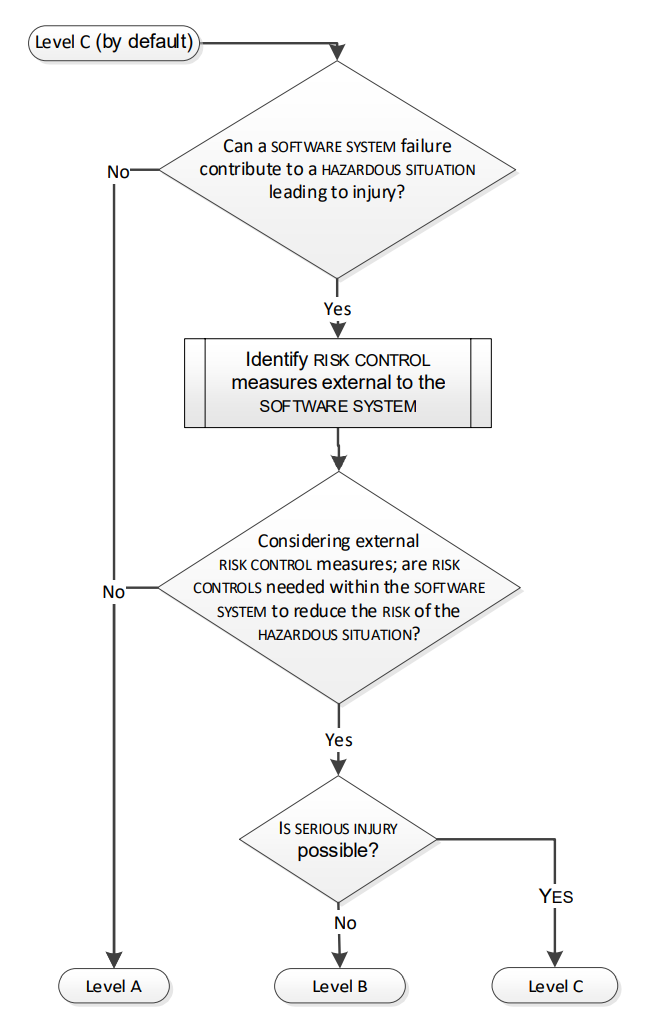
2. Weak Risk Management Integration
Risk management shall not be treated as a “binder exercise” created once during design and then filed away. Regulators expect risk management to be a living process permeating the design, fully aligning design, verification, usability, cyber security and clinical evidence.
Avoid submitting risk files that read like theoretical paperwork with little connection to how the device is actually used in the real world. As a bad example, a catheter manufacturer may list “tip breakage” as a hazard, but the clinical evaluation and V&V testing make no reference to how breakage risks were verified or mitigated. Or software documentation may claim cybersecurity risks being mitigated but provide no verification evidence of penetration testing or post-market surveillance updates.
ISO 14971 requires continuous risk evaluation, yet in practice, risks are rarely updated when design changes occur or when new field data emerges. When notified bodies or FDA reviewers see risks that don’t align with usability data, labeling, or design outputs, they quickly conclude that the risk management process is superficial.
Risk management is not a standalone FMEA document, it must be woven into the fabric of the entire Technical File.
Common issues:
- Risk analysis not reflecting real, world use
- No traceability between risks and design changes
- Incomplete hazard identification
Fix it:
- Align risk management with ISO 14971 and clinical evaluation
- Use one or more structured Hazard identification methods and update risk files with post, market surveillance (PMS) data
- Use traceability tools to link risks to mitigation
3. Poor Traceability
Traceability is the regulator’s map to understand how your device moved from concept to clinical reality. Without it, the coherence to the story of your carefully laid-out design work is broken. Are you familiar with situations like incomplete connections between risk mitigations and verification evidence: the file may list “risk of overheating mitigated by thermal cutoff,” but there’s no test showing the cutoff works across all device variants?
For FDA, these traceability gaps are red flags that the design process is not under control. For MDR, they can trigger a major finding under Annex II requirements. In one real case, a Class II manufacturer was delayed by nine months because their verification reports couldn’t be traced to the correct device configuration, making it impossible to prove the tested version matched the one in the submission.
For FDA submissions, traceability is critical. It must show a logical flow from user needs to design inputs, outputs, verification, and validation. Without it, regulators will assume the worst: that your design is uncontrolled, your risk mitigations are paper product, and your device may not be safe.
Common pitfalls:
- Missing traces between design inputs and outputs
- Missing traces between design input / output and tests
- Missing traces between design inputs and risks as well as mitigations and design inputs
Fix it:
- Establish the expected traceability matrices early
- Use traceability tools with automatic trace checking
- Audit your traceability for completeness before submission
Remember: Traceability isn’t optional, it’s the backbone of your design integrity, driving the design forward.
4. Insufficient Clinical Evaluation and PMCF Planning
Under the MDR, clinical evaluation has shifted from being a one-time hurdle to being an ongoing, evidence-driven process. Unfortunately, many manufacturers still treat it as a formality, dusting off old literature reviews or relying on shaky equivalence claims.
A classic mistake is submitting a Clinical Evaluation Report (CER) with outdated studies on a comparator device that is no longer on the market, or with no access to the full data sets behind published literature. Notified bodies are increasingly rejecting these “paper exercises,” especially when the technical, biological, and clinical equivalence is not solid.
Even worse, many companies overlook Post-Market Clinical Follow-up (PMCF) planning, assuming that general PMS activities will suffice. But MDR Article 61 requires PMCF to actively confirm safety and performance in the real world.
For instance, a company marketing an orthopedic implant with only pre-market data and no plan for long-term follow-up studies, risks having their CE certificate pulled at the first surveillance audit. Regulators want to see a continuous loop of data collection, analysis, and risk reassessment. If your clinical evaluation looks static or generic, you are signaling that your device may not withstand real-world scrutiny.
Common mistakes:
- Outdated or generic Clinical Evaluation Reports (CERs)
- No Post Market Clinical Follow-up (PMCF) plan available
- Reliance on literature without access to full comparator data
Fix it:
- Conduct systematic literature reviews and clinical studies
- Develop robust PMCF plans with measurable objectives
- Justify equivalence with technical, biological, and clinical data
5. Verification and Validation Gaps
Verification and validation are supposed to be the cornerstone of your Technical File: the hard evidence that proves your device is safe and effective. Yet, time and again, submissions are undermined by V&V documentation that is fragmented, incomplete, or outdated.
Common scenarios include test reports that summarize results in a few bullet points with no raw data, missing details about which device version was tested, or reports from third-party labs with no accreditation information.
For example, a notified body once rejected a submission for a surgical instrument because the sterilization validation was performed on an earlier prototype, not the final production version. Another manufacturer faced FDA scrutiny because their biocompatibility reports didn’t specify which device configuration was actually tested, leaving reviewers unsure whether the evidence applied to the marketed product.
If your reports don’t clearly show test conditions, acceptance criteria, and results, they will not just delay your submission; they can cast doubt on whether your device is truly ready for patients.
Common issues:
- Abbreviated or partial test reports, not stating who did the test, when it was made or which version of the test case that was executed
- No linkage to device configurations, leaving unclear which version and/or variant of the device was actually tested
- Lack of accreditation details for external labs
Fix it:
- Include full test reports clearly stating which version of the device was tested, who tested in and time of test execution
- Conduct early research on which external labs to engage, evaluate their accreditation and book your slot early









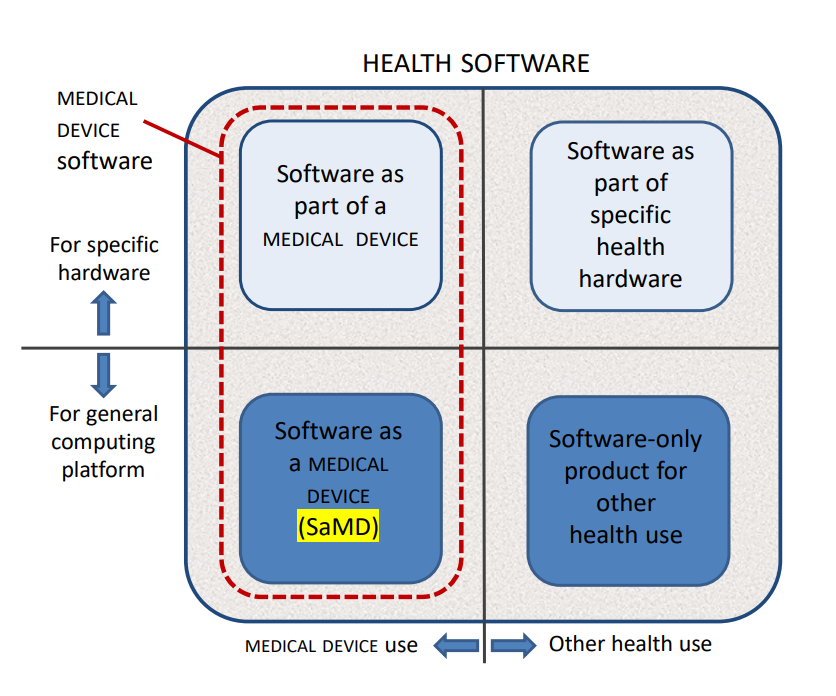 Figure 1 – HEALTH SOFTWARE field of application
Figure 1 – HEALTH SOFTWARE field of application