{fastsocialshare}
The Swiss Medtech Industry Sector study surveyed more than 340 medical device companies in this year’s SMTI industry study, uncovering the current opinions and views in the Swiss Medtech market.
The study claims that "the ever-increasing quality and documentation requirements" and the difficulty to "preserve innovative capacity" are the primary challenges to remain competitive in the current market.
For those of us working in the industry for some time, this does not come as a surprise.
Knowing the dynamics operating in medical device development, it is obvious how these two challenges inter-relate. How is it possible to sustain a competitive and innovative R&D program when more and more of the development effort is devoted to satisfying regulatory and documentation requirements?
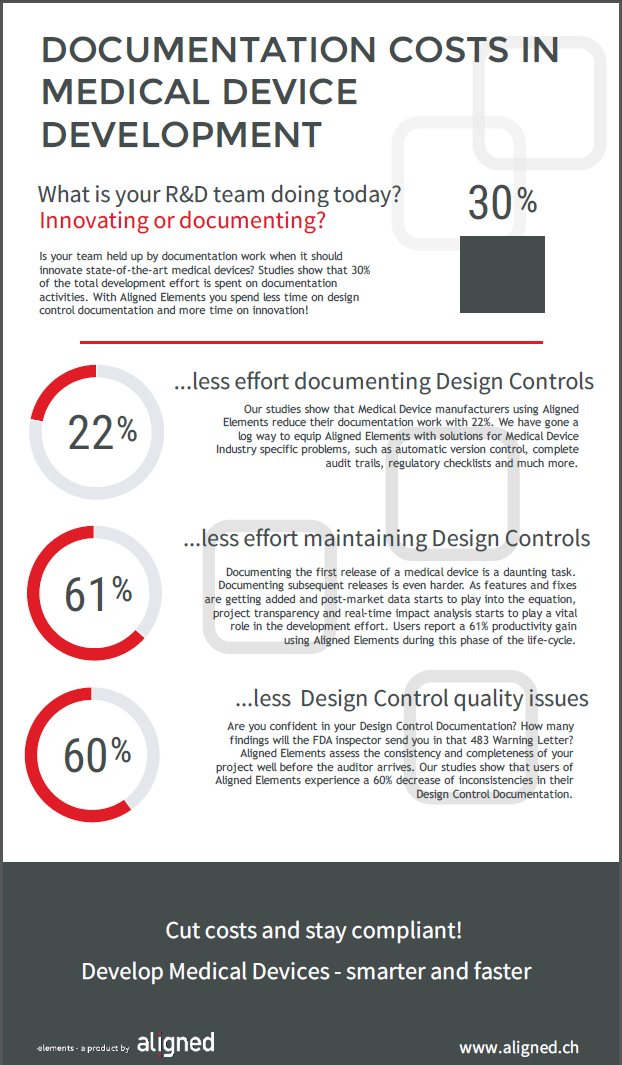
Our own studies show that up to 30% of the total medical device development effort is spent on documentation tasks. As in many other industries, the digitalization of processes offers some prospects of streamlining the medical device documentation burden.
Our users say that significant efficiency improvements can be done by applying the right tools. Download your free copy today or ask us for a free online presentation.