Aligned Elements Pre-Submission Checklist

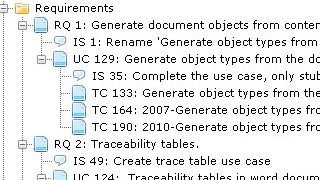
As an example of Aligned Elements project consistency control, the Aligned Elements Pre-Submission checklist demonstrates how various aspects of the project content can be analyzed in order to avoid failed audits and unwelcome FDA 483 warning letters.
Both EN/ISO 13485 and EN/ISO 14971 are concerned with the completeness and consistency of the project content and this checklist serves as an instrument to make that effort easier and faster.
The checklist guides the user through a number of analysis action steps and optionally records the results in a checklist report, which serves as objective evidence of the analysis. Detailed instructions and screenshots accompany the steps to facilitate the execution.
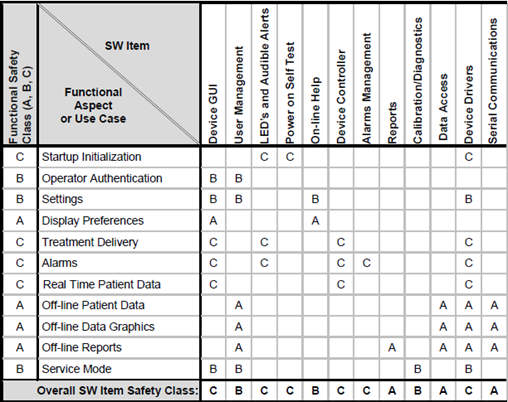
The checklist has been designed to check a project that uses the Aligned Elements default templates.
The checklist verifies test points such as:
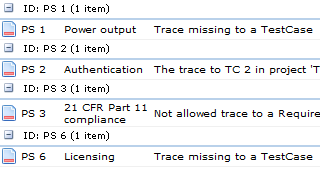
- Have all the Requirements traces to Specifications?
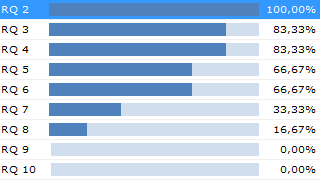
- Have all Specifications been tested?
- Are all Tests passed?
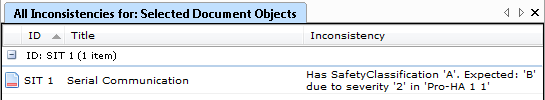
- Have all Risks been mitigated?
- Have all Mitigations been either tested or traced to a Specification?
- Have all Document Objects been placed in Word files?
- Do any Word files contain outdated Document Objects?
The checklist further performs optional checks, if you have selected to use features such as:
- The integrated Issue Management System
- The integrated Design reviews
- The integrated Risk Summary
- The integrated DHF Index
The Checklist, which is implemented as a Regulatory Wizard, is available for Aligned Elements users on the Extensions page in the Wizard section.
{fastsocialshare}