IEC 62304: Unit tests are not Unit tests

{fastsocialshare}
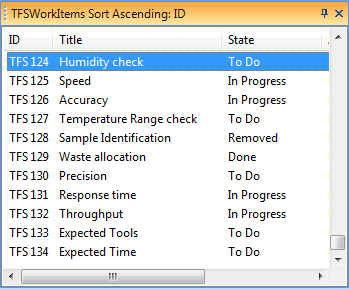
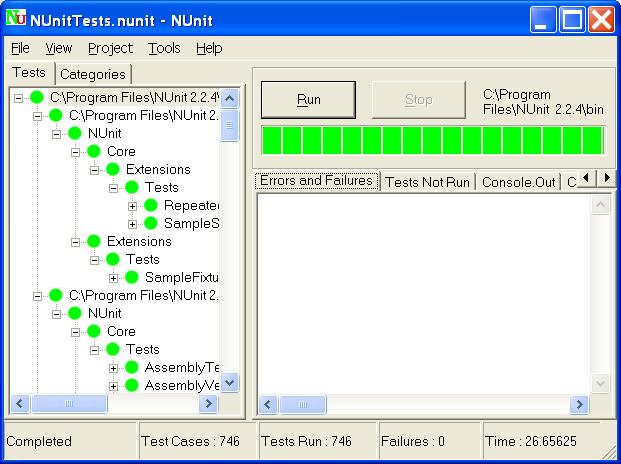
Sometimes when I ask development teams about their Software Unit verification, they fire up Nunit (or MSUnit or some other unit test tool) and say “Look here, we have more than 1800 unit tests with a test coverage way over 70%! Regarding Software Unit tests, we are certainly well covered!”
It is obvious to readers that the guys who wrote IEC 62304 explicitly tried to stay away from established software notions. You don’t see any “components”, “classes”, “modules”, “diagrams”, “dlls” or “objects” in the text (at least not in the English version). The authors did this on purpose in order to stay out of the nitty-gritty details of software development, to not make any technical prescriptions, and avoid a minefield of interpretations of concepts existing for decades.
That’s why the norm speaks of software development in tech- and methodology-neutral terms. We have “architecture”, “items”, “interfaces” and “versions”. And, unfortunately, “units”.
This is just about the only notion where there seems to be confusion. Maybe not about units per se, but certainly about “Software Units verification”, which in some companies is translated to “Unit Testing”.
What is a “Unit”?
Unit testing was popularized by the practice of Test Driven Design and extreme programming in the late ’90s. JUnit was the first xUnit testing framework getting widely used and the concept was reused for other development stacks. The xUnit testing frameworks have since then evolved and are today widely used for automated testing in many industries, including the medical device industry. Over time, the frameworks have become increasingly versatile and are today used far beyond the original scope of classical unit testing.
IEC 62304 defines the Software Unit as a Software item “not subdivided into other items”. According to the standard, it is up to the manufacturer to decide the granularity of items and therefore also the criterion for divisibility, making the definition somewhat arbitrary. Members of the medical device community have through lengthy discussions tried to agree on a practical interpretation of what a "Software Unit" is. Suggestions include:
- Class
- File
- Package
- Namespace
- Module
Unfortunately, there seems not to be a commonly accepted definition. This leaves us, the implementers of the standard, with much freedom and little support.
Regardless of the manufacturers' definition of the “Software Unit”, the standard does require the unit to be “Verified”. Again, the IEC 62304 leaves it up to the manufacturer to define according to which acceptance criteria and through which means the verification shall be conducted.
Note that the verification of a Software Unit does not necessarily have to be a test (depending on the software safety classification). The verification strategy can be a walkthrough, inspection, review, results from static testing tools, or anything else that is valid and appropriate for the Software safety classification of the unit. Furthermore, the verification strategy, methods, and acceptance criteria are permitted to vary from Unit to Unit as long as this is appropriately documented.
Let’s now move the focus from the definition of a “Software Unit” in IEC 62304 to the definition of a “Unit” in classical "Unit Testing". Wikipedia suggests several definitions (which are all variants of the word “small”) but concludes that at the business end of unit testing, we find a piece of code that can be tested in isolation. This is in general a much finer level of granularity than "Software Units" in IEC 62304. Whereas a “Software Unit” in IEC 62304 is an architectural building block, a “Unit” in Unit Testing is simply something that can be tested in isolation with no explicit relation to the software architecture.
Thus, summary so far:
- A “Unit” as in Unit Testing is not the same thing as a “Software Unit” in IEC 62304.
- A Test being executed in a xUnit Testing Framework does not automatically make it:
- a classical Unit Test
- a Software Unit Verification.
- A Software Unit can be verified by other means than tests (depending on the software safety classification).
“But I really want to use my xUnit Tests for Software Unit Verification! What do I need to do?”
It is of course entirely possible to use unit tests run by an xUnit Testing Framework (or manually if preferred) as a verification method for Software Unit. However, the tests themselves are only a part of the required deliverables. Be careful to:
- Document the test strategies, the test methods, and the procedures used.
- Evaluate the procedures for adequacy and document the results.
- Establish and document acceptance criteria (more rigid for class C than A and B, see IEC 62304)
- Perform the tests and document the result.
- Explicitly evaluate if (and document that) the results fulfill the acceptance criteria.
Learn more about how Aligned Elements can help you support IEC 62304
Request a live demo and let us show you how Aligned Elements can help you to comply to IEC 62304