Speed up your Computer Software Validation activities

{fastsocialshare}
Computer Software Validation is something we discuss a lot where I work. Since Aligned Elements is a Medical Device Quality Management System relevant software, our customers make sure that their Aligned Elements installations and configurations are validated.
It is a necessary activity, although not always perceived as bringing an awful lot of value. Sometimes the cost of CSV activities actually supersedes the cost of acquiring the software itself.
I sometimes get the question if I know any best practices when it comes to Computer Software Validation and I have made a few observations from my CSV experiences.
Let me tell you about the top 3 things that have an impact on the Computer Software Validation effort:
1) How many test cases do you perform?
It is dead simple. More test cases (note: not necessarily "more requirements") is more work. But do YOU have to perform all those test cases? What if the supplier has already verified them?
You are very much allowed to leverage existing supplier documentation and testing records. A properly conducted Supplier / Vendor assessment can lead you to the conclusion that the suppliers' verification documentation suffices. Remember that the risk we are trying to mitigate with a Computer Software Validation effort is primarily about patient safety. A software like Aligned Elements is not directly involved in patient safety and this fact can be leveraged when assessing and deciding on the validation scope.
2) How do you record the test results?
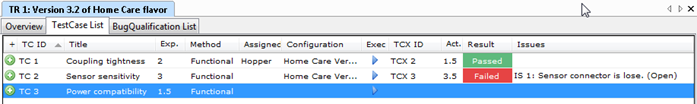
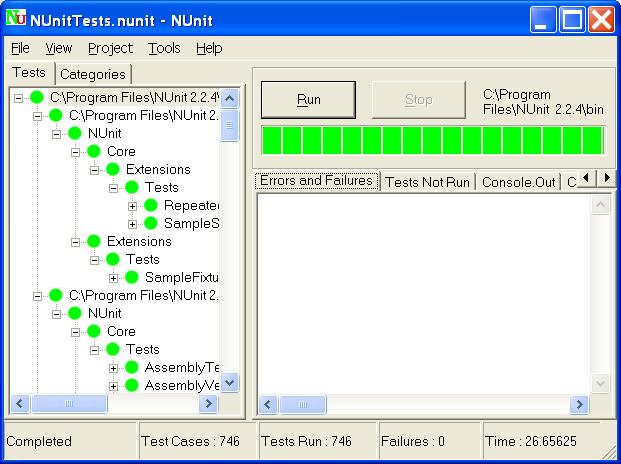
The bulk of the CSV work resides in performing and recording the test results. The way you record those results can have a significant impact on the overall effort. In order to record that the actual behavior of a test step corresponds to the expected behavior, is it enough to tick a box (passed/failed)? Or do you need to write a text? Or do you need to make screenshots? As you can imagine, there is a huge difference between the former and the latter.
"But are we not required to take screenshots?". The short answer is "No, you don't". Not if you do not think it proves anything more than the tester checking a box. FDA requires you to select your own methods and tools for quality assurance. If you have a good case for not making screenshots (which I think you have), you do not have to.
3) Who (and how many) has to sign all these CSV documents?
This might sound a bit odd but more than once I have run into cases where the validation is completed and everything that is missing is a signature from some top management figure. And now we run into a buy-in problem. If this guy has not been involved CSV approach and suddenly disagrees with how it was conducted ("BUT THERE ARE NO SCREENSHOTS?!?"), it can have a significant impact (significant as in "redo the validation").
So the lesson here is to get early buy-in from the people that sign the document. On a general level, reducing the number of signatures will speed up any documentation process. And you might want to contemplate the necessity of having the IQ, OQ, and PQ plans/reports in different documents (more documents to sign) or if you can combine them.
Validating Aligned Elements
When you acquire Aligned Elements, you get free access to our internal verification documents to use in your vendor assessments as well as pre-filled Validation documents to kick-start your validation. Contact us for more information at
What's up ahead regarding Computer Software Validation?
FDA announced that their much anticipated "Computer Software Assurance for Manufacturing, Operations, and Quality System Software" draft guidance should be out in 2019 but now it seems like it has been postponed to 2020. The new guidance is supposed to use a more agile approach, including a risk-based and value-creating perspective on CSV activities.




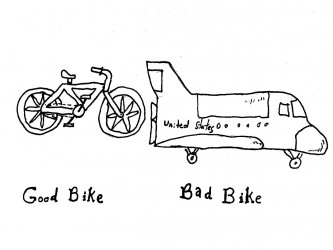 It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.
It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.